
09 / 02
2021
Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy(IF:32.621)
The microbiome of the female reproductive tract has implications for women's reproductive health.
Abstract
The microbiome of the female reproductive tract has implications for women's reproductive health. We examined the vaginal microbiome in two cohorts of women who experienced normal term births: a cross-sectionally sampled cohort of 613 pregnant and 1,969 non-pregnant women, focusing on 300 pregnant and 300 non-pregnant women of African, Hispanic or European ancestry case-matched for race, gestational age and household income; and a longitudinally sampled cohort of 90 pregnant women of African or non-African ancestry. In these women, the vaginal microbiome shifted during pregnancy toward Lactobacillus-dominated profiles at the expense of taxa often associated with vaginal dysbiosis. The shifts occurred early in pregnancy, followed predictable patterns, were associated with simplification of the metabolic capacity of the microbiome and were significant only in women of African or Hispanic ancestry. Both genomic and environmental factors are likely contributors to these trends, with socioeconomic status as a likely environmental influence.
Growing evidence suggests that the impact of the vaginal microbiome extends to the health of pregnant women and their neonates in utero and beyond. The vaginal microbiome consists of a finite number of discrete microbial communities dominated by different bacterial taxa or combinations thereof1,2. A vaginal microbiome with microbial communities dominated by species of Lactobacillus has been associated with adverse conditions of health of the female reproductive tract, whereas a microbiome dominated by complex microbial communities of Gardnerella, Atopobium, Dialister, Peptoniphilus, Lachnospiraceae members (bacterial vaginosis (BV)-associated bacterium 1 (BVAB1)) and other anaerobes3-5 has been associated with a higher risk. A complex vaginal microbiome is associated with BV, the most common gynecological condition of women of reproductive age6, as well as a higher risk of sexually transmitted infection, pelvic inflammatory disease and adverse pregnancy outcomes including preterm birth (PTB)3,5,7.
More than 10% of neonates in the United States are delivered preterm (<37 weeks’ gestation), and certain racial and ethnic groups have even higher rates8-10. Women of African ancestry in the United States are significantly more likely than women of European ancestry to have a premature birth or experience very preterm delivery (<32 weeks’ gestation). This health disparity could be due to differences in the vaginal microbiomes of these women as the population attributable risk of BV for spontaneous PTB at <32 weeks’ gestation has been estimated to be ~40%11. Although environmental factors, including socioeconomic status (for example, household income, access to care and so on) are known to contribute to these differences, genetic factors also play a role12-14. Recent studies1,2,15-23 show that the vaginal microbiomes of women of African ancestry are less likely to be dominated by species of Lactobacillus, and more likely to comprise primarily Gardnerella vaginalis, Atopobium vaginae, Sneathia amnii, BVAB1 and other anaerobes. Independent of the underlying cause, this difference is a probable contributor to the observed disparities in risk of PTB.
Here, we report an analysis of data from a collaborative effort under the umbrella of the National Institutes of Health Human Microbiome Project phase 1 (HMP1)24 and the integrative HMP (HMP2)25. In our HMP1 study, the Vaginal Human Microbiome Project (VaHMP), we collected more than 40,000 vaginal, cervical, introital and buccal swab samples cross-sectionally from a racially diverse cohort of 4,851 women visiting VCU Medical Center women’s clinics (Fig. 1a). In our HMP2 study, the Multi Omic Microbiome Study: Pregnancy Initiative (MOMS-PI), we collected more than 200,000 vaginal, buccal, rectal, birth product and other samples longitudinally from a racially diverse cohort of 1,572 women and neonates during and after their pregnancies from VCU Medical Center Clinics and clinics associated with the Global Alliance to Prevent Prematurity and Stillbirth (GAPPS) in Seattle (Fig. 1b). We present our findings from women in these two cohorts who experienced uncomplicated term birth (>37 weeks’ gestation). The samples were analyzed by 16S ribosomal RNA taxonomic profiling, metagenomic and metatranscriptomic sequencing, and metabolic pathway analysis to track dynamics of the vaginal microbiome through pregnancy. Our results show that the vaginal microbiome changes during normal pregnancy, becoming more Lactobacillus-dominated at the expense of G. vaginalis and other anaerobes. These changes are most significant in women of African ancestry, occur early in pregnancy, and seem to be the result of stabilization of Lactobacillus colonization and destabilization of colonization by other taxa. Pathway profiles generated from our metagenomic and metatranscriptomic data confirmed a transition early in pregnancy. Together, these observations suggest that the compositions of the vaginal microbiomes of women of African and non-African ancestry respond differently during pregnancy due to complex interactions between human and microbial physiology and environmental influences.

Fig. 1 ∣Overview of the VaHMP study and the MOMS-PI Term Birth study.
a, Of the 4,851 women enrolled in the VaHMP study, 613 pregnant and 1,969 non-pregnant women who reported no health complaints were analyzed in this project. A subset of 600 of these women (that is, 300 pregnant and 300 non-pregnant women, case-matched for self-reported race, gestational age at sampling and household income) was selected for analysis. Of these, there were 156 case-matched pairs of women with African ancestry, 61 case-matched pairs of women with European ancestry, and 83 case-matched pairs of women with Hispanic ancestry. These women were sampled at regular visits to VCU women’s health clinics. b, The 90 women (49 of African ancestry, 41 of European ancestry) forming the MOMS-PI Term Birth cohort were selected from a Phase 1 cohort (dark shade, pregnant women, N = 627) of women enrolled from women’s clinics at VCU. These participants were sampled longitudinally throughout pregnancy. 1st, 2nd and 3rd refer to the trimesters of pregnancy; PP, postpartum.
Results
Pregnancy alters vaginal microbiome profiles.
We generated vaginal microbiome profiles from 613 pregnant and 1,969 non-pregnant apparently healthy women from the VaHMP study as previously described15,26,27( Fig. 2a). The results suggested that pregnant women have a significantly higher (P < 0.01) prevalence of the four most common Lactobacillus vagitypes (L. crispatus, L. iners, L. gasseri and L. jensenii) and a commensurately lower prevalence of vagitypes dominated by other taxa. The primary driver of these differences was a higher (P < 0.01) prevalence of the L. iners-dominated vagitype in pregnancy at the expense of G. vaginalis and more complex vagitypes. Interestingly, incidence of BVAB1 vagitypes, a taxon associated with risk of BV and PTB7,28,29, did not decrease in pregnant women. The alpha diversity of the profiles of these pregnant women was also lower than that of non-pregnant women (P < 0.01, Fig. 1a). These observations suggest that a pregnancy-related physiological or environmental influence change in the vagina is conducive to a less complex microbiota. To further explore these observations, we compared the vaginal microbiome profiles of 300 pregnant and 300 non-pregnant case-matched women (Fig. 2b). As expected from the above-noted results, pregnant women were significantly (P < 0.01) more likely to have vagitypes of lactobacilli, with a higher predominance (P < 0.01) of the L. iners vagitype, and a commensurately lower prevalence of the G. vaginalis vagitype (P < 0.05) and other microbiome profiles. A taxon-specific analysis of these profiles confirmed a significantly higher abundance of L. iners and lower abundance of L. crispatus, G. vaginalis, A. vaginae, S. amnii, Prevotella cluster 2, Prevotella bivia and so on (Fig. 2f). Again, BVAB1 vagitypes were not affected by pregnancy. The alpha diversity of the profiles of pregnant women in this cohort was significantly lower than that of non-pregnant women (P < 0.01).
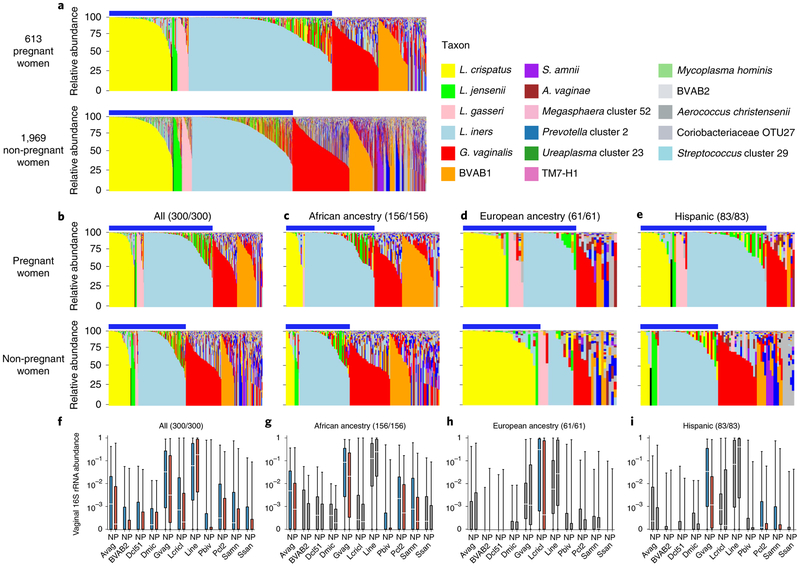
Fig. 2 ∣Pregnant and non-pregnant women of different ancestry exhibit different vaginal microbiome profiles.
a, Microbiome profiles of 613 pregnant and 1,969 non-pregnant women, 1 sample each, of all racial backgrounds taken from the VaHMP data set. Profiles were generated by 16S rRNA taxonomic classification and sorted into vagitypes representing the most dominant taxon present in the profile as previously described15,26,27. The legend is shown for a-e. Blue bars above the charts (a-e) indicate samples with Lactobacillus-dominated vagitypes. b, Microbiome profiles of a subset of 300 pregnant and 300 non-pregnant women from the VaHMP, case-matched for self-identified race, age and socioeconomic status. c, Microbiome profiles of 156 pregnant and 156 non-pregnant women of African ancestry from the VaHMP and case-matched for age and socioeconomic status. d, Microbiome profiles of 61 pregnant and 61 non-pregnant women of European ancestry from the VaHMP and case-matched for age and socioeconomic status. e, Microbiome profiles of 83 pregnant and 83 non-pregnant women of Hispanic ancestry from the VaHMP and case-matched for age and socioeconomic status. f-h, Taxon abundance differences in pregnant and non-pregnant women from b-e. Colored boxes (red and blue) indicate significant (q < 0.05 in two-sided Mann-Whitney U test after correction for false discovery rate (FDR)) differences in the abundance of that taxon between samples in women who are pregnant or not, respectively. Sample sizes for pregnant/non-pregnant cohorts are listed above each plot. Gray boxes indicate differences that are not significant. Boxes show median and interquartille range. The whiskers show the minimum and maximum values. Avag, A. vaginae; BVAB2, BV-associated bacterium 2; Dcl51, Dialister cluster 51; Dmic, Dialister micraerophilus; Gvag, G. vaginalis; Lcricl, L. crispatus cluster; Lini, L. iners; Pbiv, P. bivia; Pcl2, Prevotella cluster 2 (including P. timonensis and P. buccalis); Samn, S. amnii; Ssan, Sneathia sanguinis.
Shifts in the vaginal microbiome in pregnancy are most pronounced in women of African ancestry.
Owing to the overlap between the taxa of higher prevalence in women of African ancestry and taxa that are decreased in abundance in pregnancy, we examined the vaginal microbiome profiles of pregnant and non-pregnant women of different ancestries. Stratification of the profiles of our non-case-matched cohort according to self-reported ancestry revealed minimal differences in the microbiome profiles of pregnant and non-pregnant women of European ancestry . However, pregnant women of African and Hispanic ancestry showed a higher (P < 0.01) prevalence of vagitypes dominated by the four most prevalent Lactobacillus species than their non-pregnant counterparts. Similarly, no vagitype exhibited a significant difference in pregnant and non-pregnant women of European ancestry , and the higher prevalence of the L. iners vagitype identified in the overall cohort (Fig. 2b) was driven by the higher prevalence (P < 0.01) of this vagitype in women of African ancestry. This difference was coupled with a lower prevalence (P < 0.01) of the G. vaginalis vagitype specific to this racial group. Pregnant Hispanic women also exhibited an increased, but not quite significant, prevalence of the four Lactobacillus vagitypes. Notably, despite the lack of significant differences in the prevalence of specific vagitypes in women of European and Hispanic ancestry, the alpha diversities of the vaginal microbiomes of pregnant women in all groups were lower (P < 0.01) than those of their non-pregnant counterparts. Interestingly, the alpha diversity of women of African ancestry was consistently greater than that of women of other ancestries, independent of pregnancy status. These observations are consistent with the concept that pregnancy both favors taxa of Lactobacillus at the expense of other taxa and presents an environment conducive to a vaginal microbiome of reduced complexity.
Analysis of the case-matched cohorts of African, European and Hispanic ancestry confirmed that the differences in the profiles of the overall cohort were driven by significantly higher (P < 0.01) prevalence of Lactobacillus vagitypes with commensurately lower prevalence of other vagitypes in women of African and Hispanic ancestry (Fig. 2c,e). Women of European ancestry exhibited no significant alteration of vagitype associated with pregnancy (Fig. 2d), despite a trend toward a higher prevalence of L. iners at the expense of L. crispatus. The trends toward a higher prevalence of L. iners vagitypes and a lower prevalence of G. vaginalis vagitypes observed in the non-racially stratified group above were also observed in pregnant women of African and Hispanic ancestry, although these changes did not reach significance. All of the groups exhibited an enhanced prevalence of the L. iners vagitype in pregnancy.
A taxon-level analysis of the microbiome profiles of all women (Fig. 2f) or racially stratified groups of case-matched women demonstrated that, in women of African ancestry, G. vaginalis, A. vaginae, Prevotella cluster 2, P. bivia and S. amnii were significantly (P < 0.05) less abundant during pregnancy (Fig. 2g and Supplementary Table 2). Pregnant women of Hispanic ancestry exhibited a lower abundance of G. vaginalis, S. amnii and Prevotella cluster 2 (P < 0.05), and a lower, but not significant, abundance of A. vaginae and P. bivia (Fig. 2i ). Pregnant women of European ancestry showed only a lower abundance of L. crispatus (P < 0.05) complemented by a higher abundance of L. iners (Fig. 2h). Again, BVAB1 showed no significant change in abundance during pregnancy in these groups. The alpha diversity of the vaginal microbiomes of pregnant women in the overall case-matched cohort was significantly lower (P < 0.01). The alpha diversities of the microbiome profiles of pregnant women of African and Hispanic ancestry were also significantly lower (P < 0.05), but pregnant women of European ancestry showed a non-significant difference.
Vaginal microbiomes of women of African ancestry shift early in pregnancy.
We characterized the vaginal microbiomes in 90 pregnant women from the MOMS-PI study, 49 of African ancestry and 41 of European ancestry, who delivered at term (>37 weeks’ gestation). Overall, these women displayed an array of vagitypes reminiscent of those observed in the VaHMP study (Extended Data Fig. 3a), and the combined profiles of samples from women of African and non-African ancestry showed differences reflecting those described above (Fig. 2). Thus, pregnant women of African ancestry displayed significantly (P < 0.01) lower representation of the L. crispatus, L. gasseri and L. jensenii vagitypes, and higher (P < 0.01) representation of L. iners and BVAB1 vagitypes. Variability in women of African ancestry was driven by BVAB1 and L. iners, whereas variability in women of non-African ancestry was driven by L. crispatus and L. iners . Again, pregnancy had no significant effect on prevalence of the BVAB1 vagitype. The differences between women of African and non-African ancestry were also clearly reflected in the significantly (P < 0.01) higher alpha diversity of samples from women of African ancestry .
Samples collected longitudinally across these pregnancies suggested that pregnant women of African ancestry transition to Lactobacillus-dominated vagitypes, often to a L. iners vagitype (Fig. 3a). Prevalence of Lactobacillus-dominated profiles in women of African ancestry was lower in the first than in later trimesters, whereas women of European ancestry had a higher prevalence of Lactobacillus vagitypes throughout pregnancy. Women of African ancestry showed significantly (P < 0.05) increased prevalence of the L. iners vagitype, and a decrease of G. vaginalis and other vagitypes often associated with dysbiotic conditions. Women of non-African ancestry, who were over 75% dominated by Lactobacillus vagitypes early in pregnancy, showed no significant changes in vagitypes across pregnancy.
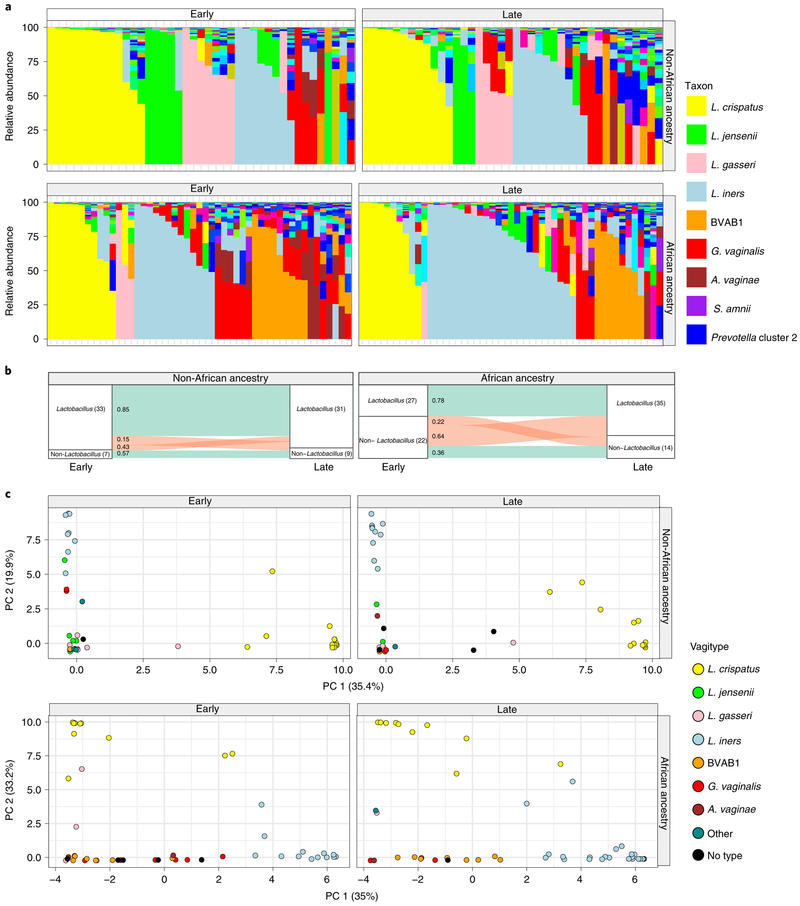
Fig. 3 ∣Vaginal microbiome profiles of women of African ancestry change early in pregnancy.
a, Vaginal microbiome profiles of pregnant women, 49 of African ancestry and 41 of non-African ancestry, collected early (the first sample before 23 weeks of gestation) and late (last sample collected after 32 weeks of gestation) during pregnancy. b, An alluvial diagram showing trajectories of transitions from Lactobacillus-dominated and non-Lactobacillus-dominated profiles of women of non-African and African ancestry across pregnancy. The number of participants in each group is indicated in brackets, with the fraction of participants transitioning indicated on the stratum. c, L1-norm principal component analysis (PCA) of samples from a. An L1-norm PCA is a method for ordination that replaces the traditional sum-of-squared errors criterion with the outlier-insensitive L1 norm54. L1-norm PCA methods capture baseline behavior in the presence of outliers when traditional PCA and principal coordinate analysis can be adversely affected.
By the second trimester, the composition of the vaginal microbiomes of pregnant women of African ancestry shows an increased prevalence of taxa of Lactobacillus driven by an increase in predominance of the L. iners vagitype (Fig. 3b,c). In the first trimester, the alpha diversity of the vaginal microbiomes of women of African ancestry was greater (P < 0.05) than that of other women, but conversion to a more Lactobacillus-dominated profile as pregnancy progressed was accompanied by a significant (P < 0.05) decrease in the alpha diversity and the number of taxa present that no longer differed significantly from that of women of non-African ancestry . In contrast, the alpha diversity of women of non-African ancestry was low and showed no significant variation throughout pregnancy (not shown). Taxon-specific analysis confirmed that, as for the cross-sectional cohort, L. iners was more abundant (P < 0.05), and G. vaginalis and A. vaginae were significantly (P < 0.05 and P < 0.01) less abundant late in pregnancy in women of African ancestry. Other taxa (for example, Prevotella cluster 2, S. amnii and TM7), following the significant trends of the more numerous participants in the cross-sectional analyses described above, were likewise less abundant late in pregnancy in women of African ancestry, although the difference did not reach significance in this smaller cohort.
Thus, the differences in the vaginal microbiomes of pregnant women are driven by changes that occur early in pregnancy in women of African ancestry. Many of the taxa decreased in pregnancy (that is, G. vaginalis, A. vaginae, Prevotella cluster 2, S. amnii and TM7) are associated with BV, pelvic inflammatory disease, sexually transmitted infections, risk of preterm birth and other adverse conditions of the female reproductive tract3,5-7.
Microbiome transitions during pregnancy follow predictable patterns.
Prevalence of a vagitype is a product of the stability of that community, the lack of stability of other vagitypes and the probabilities of switching among different dominant communities. Figure 4 shows that vagitypes dominated by lactobacilli, in particular L. crispatus, were quite stable across pregnancy. Other vagitypes (for example, G. vaginalis) exhibited less stability. Figure 4c illustrates longitudinal beta diversity measures of the samples within each participant across pregnancy. For example, samples from women with microbiomes dominated by L. crispatus at first visit are highly similar to subsequent samples collected across pregnancy, but higher instability was observed in other vagitypes (Kruskal–Wallis; P < 0.05). Stability of vagitypes examined only in women of African ancestry or in women of non-African ancestry differed but did not reach significance (not shown).

Fig. 4 ∣Temporal dynamics of vagitype transitions during pregnancy.
a, Transitions for 41 women of non-African ancestry. b, Transitions for 49 women of African ancestry. Each row represents all of the samples from a single participant, with vagitypes assigned based on 16S rRNA taxonomic profiles, shown as different color circles (see legend for code) across their pregnancies. The median Bray–Curtis dissimilarity within samples collected from the same participant is shown as a heat map to the right of each primary panel. Participants are grouped according to the vagitype of the first sample, and further sorted by decreasing median Bray–Curtis dissimilarities. c, The distribution of median Bray–Curtis dissimilarities between longitudinal samples of each women (90 women; 421 samples), grouped based on the vagitype of the first sample, is shown. Box plots were generated in R using standard approaches. The bar represents the median and the boxes indicate interquartile ranges. The whiskers show the minimum and maximum values.
Tracking of the microbiome profiles of individual women through pregnancy (Fig. 4a,b) shows that transitions occur more frequently from vagitypes often associated with dysbiotic conditions, but transitions to a vagitype dominated by a taxon poorly represented in the previous profile occurs infrequently. Thus, vagitype transitions follow predictable patterns. Most transitions away from a L. crispatus vagitype changed to the ‘no type’ or L. iners vagitype, and subsequent changes were often back to L. crispatus. The L. crispatus vagitype was stable and generally persistent in both women of African and non-African ancestry. In contrast, vagitypes dominated by L. iners, G. vaginalis and other vagitypes often associated with dysbiotic conditions were less persistent. Thus, G. vaginalis vagitypes transitioned frequently but only infrequently reverted to a G. vaginalis vagitype.
Owing to the lower numbers of women in the African and non-African groups, we collapsed the vagitypes into three groups (Lacto, L. iners and ‘other’), rationalizing that G. vaginalis and BVAB1 could group with ‘other’ taxa as they are all often associated with dysbiosis. In this model, the Lacto and L. iners vagitypes were quite stable (~75% and ~71%, respectively), but the ‘other’ group tended to remain in this higher diversity category (53%) or transition to the L. iners vagitype (34%), and only infrequently transitioned to the more protective Lacto group vagitypes (~13%). These trends held in both groups of women (Table 1c,d), but women of African ancestry with the Lacto vagitypes were sixfold more likely to transition to L. iners than women of non-African ancestry. Women of African ancestry with an L. iners or ‘other’ vagitype were also less likely (about ninefold and about sevenfold, respectively) to transition to the Lacto vagitypes. Conversely, women of African ancestry with L. iners vagitypes were threefold more likely to transition to an ‘other’ vagitype. Although these transitions failed to reach significance due to relatively low numbers, women of African ancestry underwent significantly (P < 0.05) more transitions over pregnancy than other women. Together, these observations may explain the prevalence of dysbiosis-linked vagitypes in women of African ancestry; that is, women of African ancestry seem to be less likely to transition from dysbiosis-linked vagitypes to ‘Lacto’ vagitypes and more likely to transition from ‘Lacto’ vagitypes to vagitypes more often associated with dysbiosis.
The functional potential of the vaginal microbiome changes early in pregnancy.
Metagenomic and metatranscriptomic taxonomic profiles of the vaginal microbiomes of women in this study largely recapitulated the vagitypes (Fig. 5a,,bb,,dd--f).f). However, G. vaginalis transcripts unexpectedly seemed to predominate over transcripts from BVAB1 and other taxa (Fig. 5f), suggesting that G. vaginalis is more transcriptionally active. Analysis of pathways predicted from the metagenomic and metatranscriptomic sequence data (Fig. 5c,,gg,,h)h) and analysis by sparse partial least squares discriminant analysis (PLS-DA) largely reflected the vagitypes in these samples. Whereas L. crispatus and L. iners showed discrete clusters in both metagenomic and metatranscriptomic pathway data, other vagitypes exhibited less discrete overlapping groups. Heat maps of the pathway analysis also resolved the vagitypes. A pathway centric analysis of the metagenomic data identified only ten largely unrelated pathways that significantly (P < 0.05) discriminated ancestry, likely due to the substantial partitioning of the vaginal microbiomes of women of both African and non-African ancestry into relatively discrete community states or vagitypes. However, analysis of the metatranscriptomic data identified eight pathways associated with nucleotide metabolism that were significantly (P < 0.05) less prevalent in the more complex microbial communities in women of African ancestry.
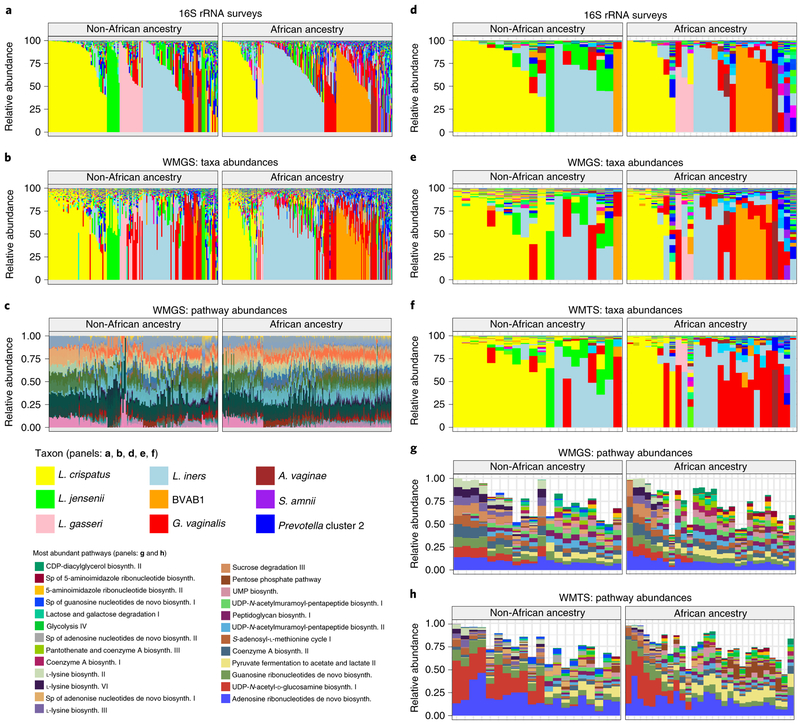
Fig. 5 ∣Metagenomic, metatranscriptomic and pathway analyses of vaginal microbiome samples support metabolic differences among vagitypes of pregnant women of African and non-African ancestry.
Longitudinal samples from 90 women, 49 of African and 41 of non-African ancestry were compared. a, 16S rRNA taxonomic assignments of samples from all three trimesters from women of non-African and African ancestry. b, Taxonomic profiles from metagenomic sequence data of samples from all three trimesters from women of non-African and African ancestry. c, Relative abundances of metabolic pathways estimated using HUMAnN255 from metagenomic sequence data of samples from all three trimesters from women of non-African and African ancestry. d, 16S rRNA taxonomic assignments of samples from the second or early third trimester (one sample per pregnancy) from women of non-African and African ancestry. e, Taxonomic profiles from metagenomic sequence data from samples as in d. f, Taxonomic profiles from metatranscriptomic sequence from samples as in d. g, Relative abundances of 25 highly abundant metabolic pathways estimated using HUMAnN2 from metagenomic sequence data from samples as in d. h, Relative abundances of 25 highly abundant metabolic pathways estimated using HUMAnN2 from metatranscriptomic sequence from samples.
Analysis of metabolic capacity of the microbiomes across trimesters showed 17 pathways significantly (P < 0.05) reduced, and 90 of 94 abundant pathways reduced in prevalence between trimesters 1 and 2. Only two pathways were significantly (P < 0.05) more reduced in prevalence between trimesters 2 and 3. Thus, the most prevalent changes in the functional microbiome occur by the first trimester. Of the 17 pathways significantly less prevalent in pregnancy, 5 were associated with protein synthesis, 5 with carbohydrate metabolism and cell wall synthesis, 4 with nucleotide metabolism, and 3 with coenzyme A and other cofactor biochemistry. Thus, pregnancy is associated with a simplification of the metabolic capacity of the resident vaginal microbiome, consistent with a transition toward a less complex Lactobacillus-dominated microbiome.
Noting a discrete pattern of pathways in L. crispatus- dominated vagitypes (Fig. 5c,,gg,,h),h), we directly compared the pathways of L. crispatus vagitypes to those of other major vagitypes. The L. iners vagitype showed the fewest significant differences (P < 0.05), mostly in pathways associated with cell wall/ membrane biochemistry. G. vaginalis, BVAB1, ‘no type’ and ‘all other’ vagitypes differed significantly (P < 0.05) in many pathways (for example, carbohydrate metabolism, cell wall/membrane biochemistry, nucleotide metabolism) from the L. crispatus vagitype. Many of the pathways (for example, carbohydrate metabolism, cell wall/envelope biochemistry, protein synthesis and nucleotide metabolism) that differed significantly (P < 0.05) between the L. crispatus vagitypes and the G. vaginalis, BVAB1, ‘no type’ and ‘all other’ vagitypes, were common. The majority of the pathways, except those associated with l-lysine biosynthesis, UDP-N-acetyl-d-glucosamine biosynthesis and acetylene degradation, were less prevalent in L. crispatus vagitypes than in other microbiome profiles. These observations are consistent with the sparse PLS-DA analysis of pathways , which showed a significant overlap of the G. vaginalis, BVAB1, ‘no type’ and ‘all other’ vagitypes.
Strains of G. vaginalis show racial and taxonomic proclivities.
Bacterial strains of the same species often exhibit significant sequence heterogeneity. G. vaginalis is now subclassified into at least four clades exhibiting nucleotide sequence divergence of ~4 to 20%30-33. Examination of our metagenomic data using PanPhlAn34 readily discriminated the four major clades of Gardnerella. G. vaginalis clade 4 was most common, but clade 3 was tightly associated with BVAB1 (P < 0.01), and was detected only in vaginal microbiomes with BVAB1 concurrently or at a previous or subsequent sampling. Although BVAB1 is found primarily in women of African ancestry, the correlation between G. vaginalis clade 3 and African ancestry was not significant, perhaps because there are many women of this ancestry whose microbiomes lack BVAB1. However, 10 of 11 women with clade 3 were of African ancestry, suggesting a linkage between this clade and ancestry. No other G. vaginalis clade showed a strong linkage to any other taxon or racial group. Since both BVAB1 and G. vaginalis are associated with higher risk of adverse reproductive health effects7,28,35-37 and both are prevalent in women of African ancestry, the apparent linkage between BVAB1 and G. vaginalis clade 3 may be relevant to racioethnic disparities in women’s reproductive health.
These analyses also revealed discrete but previously uncharacterized clades of L. crispatus, L. jensenii, L. gasseri, L. iners and BVAB1. We observed no relationship between these clades and any other parameter, although, strain-level discrimination is likely relevant to understanding the impact of these microbial communities on human health.
Discussion
Our VaHMP and MOMS-PI cohorts of women of European, African and Hispanic ancestry exhibit different vaginal microbiome compositions and dynamics during pregnancy. The cause of these differences remains unclear, although both genetic and environmental influences are likely contributors. Potential confounding factors that our analyses have yet to resolve include socioeconomic status and age. Thus, in our cohorts, women of African and Hispanic ancestry are significantly younger and have lower household income than women of European ancestry. Our previous analysis15 of non-pregnant women indicated that race has a stronger association with diversity of the vaginal microbiome than socioeconomic status. Thus, although we were unable to completely decouple race from socioeconomic status herein, we believe both have an impact.
Our longitudinal analysis argues that the transitions of the vaginal microbiomes occur early in pregnancy. Stout et al.38 reported a decrease in richness and diversity early in pregnancy in the vaginal microbiomes of predominantly African American women who would later deliver preterm. However, in that study of only 149 longitudinal samples from 77 women, no change was observed in women who would deliver at term. DiGiulio et al.39 reported a stable pregnancy microbiome of the gut, saliva and mouth, and in contrast to results reported herein, the vagina. The cohort of women in that project was predominantly (22 of 34) self-reported as ‘White’ with only 1 woman self-reported as ‘Black’. Goltsman et al.40, following ten white pregnant women (six who delivered term and four who delivered preterm), presented data that suggest stability of the vaginal microbiome in pregnancy, although four of the women with vaginal microbiomes initially dominated by L. iners showed an increase in diversity with gestation. Together, these results are largely consistent with our observation that vaginal microbiome changes are most significant in women of African ancestry.
It appears that changes occurring during pregnancy may render the reproductive tracts of women of all racial backgrounds more hospitable to taxa of Lactobacillus and less favorable for G. vaginalis and other taxa associated with BV and dysbiosis. Since the changes are generally toward a microbiome composition resembling that of non-pregnant women of European ancestry, it is not surprising that the shifts are more striking in women of African and Hispanic ancestry. As we and others have speculated7,16,17,41,42, hormonal and other physiological changes in pregnancy may promote a vaginal environment conducive to a less dysbiotic vaginal microbiome. We previously demonstrated43 that non-pregnant women taking combined oral contraceptives have vaginal microbiome profiles with a higher predominance of Lactobacillus taxa, and several studies by others44-47 have shown that post-menopausal women receiving hormone replacement therapy tend to have a more Lactobacillus-dominated microbiome. Elevated estrogens in pregnancy enhance glycogen synthesis in the vaginal epithelium. Although most lactobacilli lack alpha-amylase, human enzymes and other bacterial pathways process glycogen into compounds (for example, maltose, maltotriose, maltopentaose and maltodextrins) that are fermented by these bacteria48. Thus, Lactobacillus are apparently selectively enriched, to the detriment of other microbial taxa, in pregnant women. Interestingly, BVAB1, which has been associated with dysbiotic vaginal conditions and risk of PTB7,28,29, and which is present as a major vagitype largely in women of African ancestry, is not noticeably decreased in prevalence in pregnancy. Thus, BVAB1, for reasons yet to be determined, is apparently resistant to factors sculpting the microbiome in pregnant women, possibly explaining in part the enhanced risk for PTB experienced by women of African ancestry.
We and others1,2,15-23 have previously shown that vaginal microbiomes of non-pregnant women of European ancestry are less complex than those of non-pregnant women of African ancestry. However, the differential impact of pregnancy on the vaginal microbiomes of women of diverse ancestry has not been comprehensively explored. An early cross-sectional study by Aagaard et al.49 probed the microbiomes of 24 pregnant and 60 non-pregnant women and found that, in pregnancy, the vaginal microbiome is reduced in complexity and enriched in species of Lactobacillus. Walther-Antonio et al.41 and Romero et al.42 followed 8 pregnant women and 22 pregnant/32 non-pregnant women, respectively, and also concluded that lactobacilli dominate the vaginal microbiome during pregnancy. MacIntyre et al.16, in a longitudinal study of a British cohort of 42 pregnant women who experienced term birth, and Freitas et al.50 in a cross-sectional study of 182 pregnant and 310 non-pregnant Canadian women, reported reduced diversity and higher prevalence of lactobacilli in vaginal microbiomes during pregnancy. Although these studies are consistent with the results reported herein, numbers of participants were quite modest, non-pregnant case-matched controls were often not available, taxon-specific changes other than Lactobacillus species were generally not addressed, and variables associated with race and ethnicity were usually not explored.
In our study, taxa that are decreased in prevalence in pregnancy include G. vaginalis, A. vaginae, S. amnii, Prevotella cluster 2, P. bivia and others. Many of these taxa have been associated with adverse health conditions, including BV, susceptibility to sexually transmitted infections and human immunodeficiency virus, and adverse pregnancy outcomes including preterm birth7,28,35-37. Thus, our results are suggestive of a relationship between physiological changes occurring during pregnancy and the development of a less dysbiotic vaginal microbiota (that is, a more disease-resistant state).
Our examination of the probability of transitions among vagitypes provides clues to the reasons that women of African ancestry generally have more complex vaginal microbiome profiles. Thus, the more favorable Lactobacillus vagitypes (that is, the L. crispatus, L. jensenii and L. gasseri vagitypes) are more stable, and transition to those vagitypes is less common in women of African ancestry. In contrast, vagitypes associated with dysbiosis (BVAB1, G. vaginalis, A. vaginae, S. amnii and other vagitypes) are less stable and transition to other vagitypes at a higher rate. Moreover, transitions from dysbiotic vagitypes to favorable vagitypes are more frequent in women of non-African ancestry. As a result, women of African ancestry are more likely to have a dysbiotic vagitype than women of non-African ancestry. These observations are reminiscent of Gajer et al.17, who in a study of 32 non-pregnant reproductive age women reported that vaginal communities dominated by Lactobacillus species were quite stable in contrast to more complex vaginal microbiomes, although that report did not focus on differences in transition rates associated with ancestry or pregnancy. The fact that the directions of these microbiome transitions are similar in pregnant and non-pregnant women suggests that the forces driving them may be related.
Metagenomic analysis illustrated minimal differences in metabolic potential associated with the overall vaginal microbiomes of women of African and non-African ancestry. In contrast, our metatranscriptome analysis identified pathways associated with nucleotide metabolism that are less prevalent in women of African ancestry. This result is consistent with a hypothesis suggesting that a more complex microbiome may be associated with greater damage to the vaginal epithelium, which may release metabolic precursors (for example, nucleotides and nucleotide precursors) that could be scavenged by the vaginal microbiota. Interestingly, nucleotide biosynthesis has been identified as a critical capability for bacterial proliferation in the human bloodstream51. It would not be surprising if this were also true in other niches of the human body.
Metagenomic and metatranscriptomic data permitted subspecies classifications. Thus, four previously identified clades of G. vaginalis30-33 and two new clades each of L. crispatus, L. jensenii, L. gasseri, L. iners and BVAB1 were discriminated. Clade 3 of G. vaginalis was tightly associated with BVAB1, suggesting a synergistic or symbiotic relationship. Although BVAB1 is highly correlated with women of African ancestry, the relationship between clade 3 of G. vaginalis and race was not significant, probably because many women of African ancestry have microbiome profiles dominated by other taxa. Recently, Janulaitiene et al.52, studying a population of 109 non-pregnant women with and without BV, found, as did we, that clade 4 was most common, followed by clade 3, and identified a correlation between clades 1 and 2 and a high Nugent score indicative of BV. Callahan et al.53, studying a cohort of 39 pregnancies, 9 of which ended prematurely (<37 weeks’ gestation), showed data suggesting an association between a G. vaginalis group, termed G3, that includes isolates from both clades 1 and 2, and preterm birth. We have not yet detected a correlation between clades 1, 2 or 4 and any other taxon or clinical condition. However, these observations of close linkage between specific microbial taxa and clinical and demographic phenotypes are suggestive of interdependencies that remain to be elucidated.
The fact that some groups frequently have vaginal microbiome profiles that are clearly associated with dysbiosis, BV and risk of adverse outcomes in pregnancy, including but not limited to preterm birth, underscores the compelling need to further explore these phenomena. That the vaginal microbiomes known to confer higher risk of poor health and adverse outcomes of pregnancy are more highly associated with women of African and Hispanic ancestry, but that pregnancy tends to drive these microbiomes toward more favorable microbiota, suggests that an external intervention that favors this trend might be beneficial for these populations. Characterization of the composition of the vaginal microbiome is now readily achieved. What remains is to verify the most favorable microbiome and the most effective strategy for intervention.
References
1. Ravel J et al. Vaginal microbiome of reproductive-age women. Proc. Natl Acad. Sci. USA 108, 4680–4687 (2011).
2. Ravel J et al. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome 1, 29 (2013).
3. Younes JA et al. Women and their microbes: the unexpected friendship. Trends Microbiol. 26, 16–32 (2017).
4. Srinivasan S et al. Temporal variability of human vaginal bacteria and relationship with bacterial vaginosis. PloS One 5, e10197 (2010).
5. Petrova MI, Lievens E, Malik S, Imholz N & Lebeer S Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front. Physiol 6, 81 (2015).
6. Sobel JD Bacterial vaginosis. Annu. Rev. Med 51, 349–356 (2000).
7. Fettweis JM et al. The vaginal microbiome and preterm birth. Nat. Med 10.1038/s41591-019-0450-2 (2019).
8. Shah R et al. Incidence and risk factors of preterm birth in a rural Bangladeshi cohort. BMC Pediatr. 14, 112 (2014).
9. Tielsch JM Global incidence of preterm birth. Nestle Nutr. Inst. Workshop Ser 81, 9–15 (2015).
10. WHO. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity Bull. World Health Org; 88, 31–8 (2010).
11. Goldenberg RL et al. The preterm prediction study: the value of new vs standard risk factors in predicting early and all spontaneous preterm births. NICHD MFMU Network. Am. J. Public Health 88, 233–238 (1998).
12. York TP, Eaves LJ, Neale MC & Strauss JF The contribution of genetic and environmental factors to the duration of pregnancy. Am. J. Obstet. Gynecol 210, 398–405 (2014).
13. Barcelona de Mendoza V et al. A systematic review of DNA methylation and preterm birth in African American women. Biol. Res. Nurs 19, 308–317 (2017).
14. Modi BP et al. Mutations in fetal genes involved in innate immunity and host defense against microbes increase risk of preterm premature rupture of membranes (PPROM). Mol. Genet. Genom. Med 5, 720–729 (2017).
15. Fettweis JM et al. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiolology 160, 2272–2282 (2014).
16. MacIntyre DA et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep 5, 8988 (2015).
17. Gajer P et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med 4, 132ra52 (2012).
18. Hyman RW et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci 21, 32–40 (2014).
19. Ma B, Forney LJ & Ravel J The vaginal microbiome: rethinking health and diseases. Annu. Rev. Microbiol 66, 371–389 (2012).
20. Hickey RJ, Zhou X, Pierson JD, Ravel J & Forney LJ Understanding vaginal microbiome complexity from an ecological perspective. Transl. Res. J. Lab. Clin. Med 160, 267–282 (2012).
21. Martin DH & Marrazzo JM The vaginal microbiome: current understanding and future directions. J. Infect. Dis 214, S36–S41 (2016).
22. Zhou X et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 1, 121–133 (2007).
23. Beamer MA et al. Bacterial species colonizing the vagina of healthy women are not associated with race. Anaerobe 45, 40–43 (2017).
24. Peterson J et al. The NIH Human Microbiome Project. Genome Res. 19, 2317–2323 (2009).
25. The Integrative Human Microbiome Project. Dynamic analysis of microbiome–host omics profiles during periods of human health and disease. Cell Host Microbe 16, 276–289 (2014).
26. Fettweis JM et al. Species-level classification of the vaginal microbiome. BMC Genom. 13, S17 (2012).
27. Brooks JP et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 15, 66 (2015).
28. Nelson DB et al. Early pregnancy changes in bacterial vaginosis-associated bacteria and preterm delivery. Paediatr. Perinat. Epidemiol 28, 88–96 (2014).
29. Fredricks DN, Fiedler TL, Thomas KK, Oakley BB & Marrazzo JM Targeted PCR for detection of vaginal bacteria associated with bacterial vaginosis. J. Clin. Microbiol 45, 3270–3276 (2007).
30. Lopes dos Santos Santiago G et al. Gardnerella vaginalis comprises three distinct genotypes of which only two produce sialidase. Am. J. Obstet. Gynecol 204, 450.e1–7 (2011).
31. Piot P et al. Biotypes of Gardnerella vaginalis. J. Clin. Microbiol 20, 677–679 (1984).
32. Ingianni A, Petruzzelli S, Morandotti G & Pompei R Genotypic differentiation of Gardnerella vaginalis by amplified ribosomal DNA restriction analysis (ARDRA). FEMS Immunol. Med. Microbiol 18, 61–66 (1997).
33. Ahmed A et al. Comparative genomic analyses of 17 clinical isolates of Gardnerella vaginalis provide evidence of multiple genetically isolated clades consistent with subspeciation into genovars. J. Bacteriol 194, 3922–3937 (2012).
34. Segata N et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814 (2012).
35. Onderdonk AB, Delaney ML & Fichorova RN The human microbiome during bacterial vaginosis. Clin. Microbiol. Rev 29, 223–238 (2016).
36. Menard JP et al. High vaginal concentrations of Atopobium vaginae and Gardnerella vaginalis in women undergoing preterm labor. Obstet. Gynecol 115, 134–140 (2010).
37. Eastment MC & McClelland RS Vaginal microbiota and susceptibility to HIV. AIDS Lond. Engl 32, 687–698 (2018).
38. Stout MJ et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am. J. Obstet. Gynecol 208, 226.e1–7 (2013).
39. DiGiulio DB et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl Acad. Sci. USA 112, 11060–11065 (2015).
40. Goltsman DSA et al. Metagenomic analysis with strain-level resolution reveals fine-scale variation in the human pregnancy microbiome. Genome Res. 28, 1–14 (2018).
41. Walther-António MRS et al. Pregnancy’s stronghold on the vaginal microbiome. PloS One 9, e98514 (2014).
42. Romero R et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2, 4 (2014).
43. Brooks JP et al. Effects of combined oral contraceptives, depot medroxyprogesterone acetate and the levonorgestrel-releasing intrauterine system on the vaginal microbiome. Contraception 95, 405–413 (2016).
44. Muhleisen AL & Herbst-Kralovetz MM Menopause and the vaginal microbiome. Maturitas 91, 42–50 (2016).
45. Cauci S et al. Prevalence of bacterial vaginosis and vaginal flora changes in peri- and postmenopausal women. J. Clin. Microbiol 40, 2147–2152 (2002).
46. Pabich WL et al. Prevalence and determinants of vaginal flora alterations in postmenopausal women. J. Infect. Dis 188, 1054–1058 (2003).
47. Hillier SL & Lau RJ Vaginal microflora in postmenopausal women who have not received estrogen replacement therapy. Clin. Infect. Dis 25, S123–126 (1997).
48. Spear GT et al. Human α-amylase present in lower-genital-tract mucosal fluid processes glycogen to support vaginal colonization by. Lact. J. Infect. Dis 210, 1019–1028 (2014).
49. Aagaard K et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PloS One 7, e36466 (2012).
50. Freitas AC et al. The vaginal microbiome of pregnant women is less rich and diverse, with lower prevalence of Mollicutes, compared to non-pregnant women. Sci. Rep 7, 9212 (2017).
51. Samant S et al. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLOS Pathog. 4, e37 (2008).
52. Janulaitiene M et al. Prevalence and distribution of Gardnerella vaginalis subgroups in women with and without bacterial vaginosis. BMC Infect. Dis 17, 394 (2017).
53. Callahan BJ et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl Acad. Sci. USA 114, 9966–9971 (2017).
54. Brooks JP, Dulá JH & Boone EL A pure L1-norm principal component analysis. Comput. Stat. Data Anal 61, 83–98 (2013).
55. Franzosa EA et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962 (2018).


