
09 / 02
2021
The vaginal microbiome and preterm birth(IF:32.621)
Maternal and fetal genetics, and gene-environment interactions, clearly play roles in determining the length of gestation
Approximately 15 million preterm births at less than 37 weeks of gestation occur annually worldwide1. Preterm birth (PTB) remains the second most common cause of neonatal death across the globe, and the most common cause of infant mortality in middle- and high-income economies2.The incidence of preterm birth exceeds 10% worldwide. The consequences of PTB persist from early childhood into adolescence and adulthood3,4. In the United States, striking population differences with respect to PTB exist, with women of African ancestry having a substantially larger burden of risk. The estimated annual cost of PTB in the United States alone is over US$26.2 billion5. Despite these statistics, there remains a paucity of effective strategies for predicting and preventing PTB.
Although maternal and fetal genetics, and gene–environment interactions, clearly play roles in determining the length of gestation, environmental factors, including the microbiome, are the most important contributors to PTB, particularly among women of African ancestry6. Microbe-induced inflammation resulting from urinary tract infection, sexually transmitted infections, including trichomoniasis, or bacterial vaginosis is thought to be a cause of PTB7,8. Ascension of microbes7,9 from the lower reproductive tract to the placenta, fetal membranes and uterine cavity, and hematogenous spread of periodontal pathogens from the mouth, have also been invoked to explain the up to 40–50% of preterm births that are associated with microbial etiologies10,11.
A homogeneous Lactobacillus-dominated microbiome has long been considered the hallmark of health in the female reproductive tract. In contrast, a vaginal microbiome with high species diversity, as observed with bacterial vaginosis, has been associated with increased risk for acquisition and transmission of sexually transmitted infections, PTB and pelvic inflammatory disease12–15. However, many asymptomatic healthy women have diverse vaginal microbiota. More refined approaches are needed to assess risk, promote health, and prevent and treat disease16–21.
Recent reports of the microbiome in pregnant women22–39 have suggested that the composition of the vaginal microbiome has a significant population-specific impact on PTB risk. Several studies that focused on populations predominantly of European descent22–25 have associated Lactobacillus crispatus with a lower risk of PTB, and the finding was replicated in a cohort of predominantly African descent25. As first reported by Ravel et al.16, and subsequently confirmed in other studies21,40, the vaginal microbiome profiles of women of African and European ancestry differ significantly. Although distinct taxa have been associated with PTB in women of African ancestry in some studies25,26, others have not found significant associations27,30. Women of African descent are less likely to exhibit vaginal lactobacilli, frequently have vaginal L. crispatus predominance and are more likely to exhibit increased vaginal microbial diversity16,21. Consequently, population-specific studies may be required to assess the broad impacts of the vaginal microbiome on risk of PTB and to identify contributing taxa that may be carried by only a small subset of women.
In the present study, we report a community resource that includes samples collected longitudinally during 1,572 pregnancies of women from diverse ancestries, and omics data generated from samples collected from 597 pregnancies in a collaborative effort under the umbrella of the National Institutes of Health’s integrative Human Microbiome Project (iHMP)41. Furthermore, we provided an analysis of the longitudinal, comprehensive, multi-omic profiling of vaginal samples from 45 women who experienced spontaneous PTB and 90 case-matched controls, in a cohort of women of predominantly African ancestry. In an initial analysis of this dataset, which represents one of the largest and most comprehensive studies of the vaginal microbiome to date, we identified vaginal microbial signatures in women who went on to experience PTB.
Results
The Multi-Omic Microbiome Study: Pregnancy Initiative
The longitudinal iHMP study, the Multi-Omic Microbiome Study: Pregnancy Initiative (MOMS-PI) includes a total of 1,572 pregnancies, with 992 pregnancies from clinics associated with the Research Alliance for Microbiome Science (RAMS) Registry, based at Virginia Commonwealth University (VCU) in Virginia, and 580 pregnancies from sites associated with the Global Alliance to Prevent Prematurity and Stillbirth (GAPPS) in Washington State. The resource features two comprehensive datasets of integrated microbiome and host functional properties measured longitudinally in pregnancy and the perinatal period (Fig.1): (1) the MOMS-PI Preterm Birth (PTB) study dataset generated from a case–control study of 45 women predominantly of African ancestry, who delivered spontaneously preterm, and 90 case-matched women who delivered at term; and (2) the MOMS-PI Term Birth (TB) study dataset generated from an ethnically diverse retrospective cohort study of 90 women, who delivered at term or early term42. From a selection of 12,039 samples from 597 pregnancies, we generated: (1) 16S ribosomal RNA (rRNA) taxonomic profiles from 6,452 samples from pregnant women and 2,753 samples from neonates; (2) metagenome profiles from 930 samples from pregnant women and 146 samples from neonates; (3) metatranscriptome profiles from 297 samples from pregnant women; (4) cytokine profiles from 1,223 samples from pregnant women and 173 samples from neonates; and (5) lipid profiles from 63 samples from pregnant women. In the overall MOMS-PI study, we collected a total of 206,437 samples from pregnant women and their neonates, which have been archived in the RAMS Registry (see ramsregistry.vcu.edu) (Fig.1c). Comprehensive health history and outcome data were also collected longitudinally.
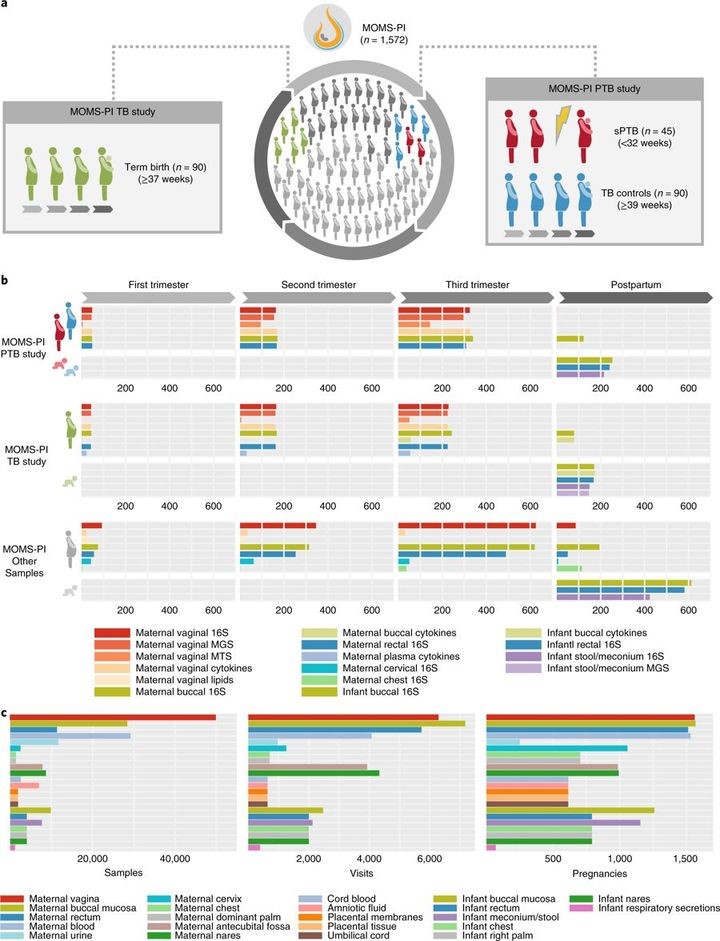
Fig. 1 MOMS-PI resources.
a, An overview of the study designs for the MOMS-PI PTB study (45 spontaneous preterm (sPTB) cases and 90 term controls) and the MOMS-PI TB study (90 women who delivered at term or early term and their neonates). Both cohorts were selected from the phase 1 RAMS Registry cohort (n = 627). b, Omics data were generated from samples from the MOMS-PI PTB and MOMS-PI TB studies and 384 additional pregnancies from the overall MOMS-PI cohort. Samples from the 12 women who were selected for both the MOMS-PI PTB study and the MOMS-PI TB study are depicted under both studies. Omics data types include 16S rRNA amplicon sequencing, metagenomic sequencing (MGS), metatranscriptomic sequencing (MTS), host cytokine assays and lipidomics. c, A total of 206,437 samples were collected at more than 7,000 visits from 1,572 pregnancies in the MOMS-PI study, and are archived in the RAMS Registry.
Vaginal microbiome profiles show PTB-associated trends
In the present study, we focus our analysis on a comprehensive multi-omic profiling of vaginal samples in the MOMS-PI PTB study. We analyzed 45 single gestation pregnancies that met the criteria for spontaneous PTB (23–36 weeks 6 days of gestational age) and 90 single gestation pregnancies that extended through term (≥39 weeks) to avoid issues possibly associated with early term births43–45.
The cohort predominantly comprised women of African ancestry (~78%), with a median annual income of less than US$20,000 and an average age of 26 years. Microbiome profiles of the first vaginal samples collected at study enrollment (Fig. 1) were generated by 16S rRNA taxonomic analysis. For vaginal samples, the dominant bacterial taxon is one clinically meaningful measure by which to stratify samples16,46. Women who went on to deliver at term were more likely to exhibit L. crispatus predominance in the vaginal microbiome (P = 0.014, Fig. 2a,b, Extended Data Fig1), paralleling earlier observations17,22–25. A Markov chain analysis to assess vagitype changes throughout pregnancy did not reveal statistically significant differences in transition rates between case and control groups.
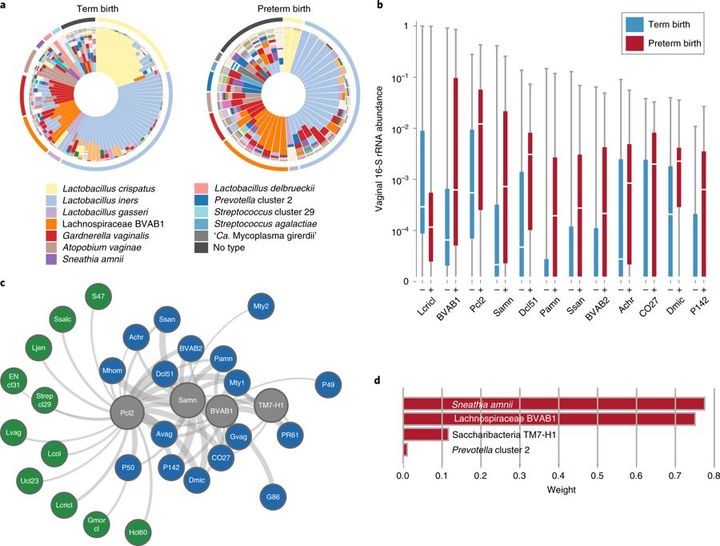
Fig. 2 Bacterial taxa associated with spontaneous PTB.
a, Vagitypes of 90 women who delivered at term (≥39 weeks of gestation), and 45 women who delivered prematurely (<37 weeks of gestation) showing 13 community states, or vagitypes. b, Abundance of taxa significantly different in PTB (n = 45) and TB (n = 90) cohorts. These taxa have P <0.05 for the Mann–Whitney U-test (two-sided) for difference in proportional abundance between the cohorts, corrected using the Benjamini–Hochberg procedure with an FDR of 5%. TB is indicated in blue as (–) and PTB in red as (+). Boxes show the median and interquartile range; whiskers extend from minimum to maximum values within each cohort. c, Network analysis of four taxa highly associated with PTBs. Negative correlations are shown in green, positive correlations in blue and predictive taxa in gray. Edge weights represent the strength of correlation. See Supplementary Table 3 for abbreviations. d, Predictive linear model for PTBs that produces a score based on weighted log(abundances) of four taxa in vaginal 16S rRNA profiles in the 6- to 24-week gestational age range. Taxa abbreviations: Lcricl, L. crispatus cluster; BVAB1, Lachnospiraceae BVAB1; Pcl2, Prevotella cluster 2; Samn, S. amnii; Dcl51, Dialister cluster 51; Pamn, P. amnii; BVAB2, Clostridiales BVAB2; CO27, Coriobacteriaceae OTU27; Dmic, Dialister micraerophilus; P142, Parvimonas OTU142.
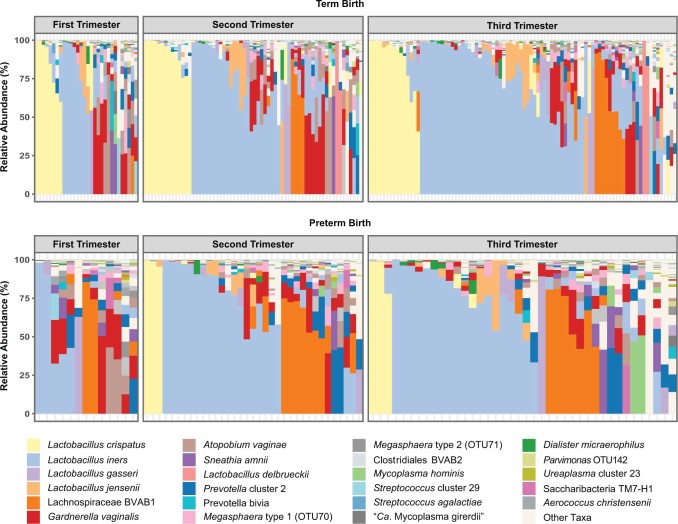
Extended Data Figure 1
Species-level vaginal microbiome composition in women who experience TB or PTB.
Stacked bar charts illustrating the vaginal microbiome profiles from 16S rRNA surveys of one sample per trimester from each pregnancy. Samples are ordered according to decreasing relative abundance. Twenty-nine abundant taxa of interest are shown with all others pooled into ‘Other’.
Early prediction of risk for PTB is critical for the development of new strategies for prevention and intervention. As a proof of concept, we developed a model for identifying the most discriminative taxa for PTB using 16S rRNA data from samples collected at 24 weeks of gestation or earlier. Model construction involved selecting taxa that are differentially represented in the cohorts as assessed using the Mann–Whitney U-test (Fig.2d), and assigning weights to these taxa using L1-regularized logistic regression. The resulting model incorporates four taxa: S. amnii, BVAB1, Prevotella cluster 2 and TM7-H1, which are all positively correlated with PTB (Fig.2d). The discriminative model is significant (P = 0.0024) and has an expected sensitivity of 77.4%, specificity of 76.3%, and an area under the receiver operating characteristics (AUROC) curve of 0.723 for samples not used during training. This model, based on microbiome composition data, had 5–7% greater sensitivity and specificity than a model constructed using only clinical variables with a slight reduction in the AUROC curve (that is, 0.723 versus 0.764). A network analysis of these four taxa (Fig.2c) shows them to be positively correlated with taxa associated with vaginal dysbiosis.
Metagenomic assembly of reference genomes of bacterial taxa associated with PTB
MGS data generated with Pacific BioSciences and Illumina sequencing technologies were used to generate the first genomes of TM7-H1 (CP026537) and BVAB1 (PQVO000000), respectively. BVAB1, with a genome of ~1.45 megabases (Mb), is classified to the Family Lachnospiraceae of the Order Clostridiales, and is not closely related to any other known bacterium. TM7-H1, with a genome of ~0.72 Mb, falls into the Phylum Candidatus Saccharibacteria and exhibits only ~66% nucleotide identity with the recently described oral TM7x isolate (NZ_CP007496)52. TM7-H1 encodes a putative α-amylase and is predicted to be able to utilize glycogen as a carbon source. Similar to TM7x52, TM7-H1 lacks de novo biosynthetic capabilities for essential amino acids, and likely depends on other organisms in the vaginal environment for survival. However, although TM7x is an obligate parasitic epibiont, it remains unknown whether TM7-H1 similarly lives on the surface of another bacterial species in the vaginal environment. We identified 243 and 421 metabolic reactions, respectively, in TM7-H1 and BVAB1. Both organisms are predicted to have the ability to produce pyruvate, acetate, l-lactate and propionate. BVAB1 encodes additional pathways for production of acetaldehyde, d-lactate, formate and acetyl-CoA. Neither is predicted to have a functional tricarboxylate cycle, and TM7-H1 completely lacks genes related to butyrate metabolism. As described above, production of short-chain fatty acids has been linked to a proinflammatory state51, with possible implications for disease.
Bacterial taxa associated with PTB in metagenomic and metatranscriptomic data
On average, approximately 95% of MGS reads and 30% of MTS reads were identified as human. Most non-human MGS and MTS reads mapped to our customized vaginal bacterial database, with only a small fraction remaining unmapped (that is, average of 0.45% full-term metagenomics, 0.41% preterm metagenomics, 1.67% full-term metatranscriptomics and 2.46% preterm metatranscriptomics) . We compared the relative proportional abundance of taxa from the 16S rRNA assay with the relative proportional abundance of metagenomic and metatranscriptomic data that mapped to non-ribosomal genes across the 56 taxa in our database. Although proportional differences were observed across detection methods, there was concordance in detection of taxa using 16S rRNA profiles, MGS and MTS.
Using the same approach with MGS data, we observed similar trends, but fewer genes were identified as statistically significant (Padj < 0.05) overall. Interestingly, 12.55% of the G. vaginalis genes analyzed using MGS data were significantly higher in the term cohort, whereas only one gene (that is, 0.02% of genes analyzed) that was identified as a hypothetical protein was higher (Padj < 0.05) in the preterm cohort. We found the overall relative transcriptional rate of G. vaginalis was higher in preterm samples compared with term samples, using a calculated ratio of the proportion of reads mapped to genes in G. vaginalis reference genomes by MTS to reads mapped by MGS (Wilcoxon’s, P < 0.05). Previous studies have suggested that PTB risk differs with carriage of different G. vaginalis clades25. We extend this finding by showing data suggesting that PTB risk also differs with the transcriptional activity of G. vaginalis. Further investigations will be required to determine the underlying mechanisms that impact replication and transcription of different strains of G. vaginalis during pregnancy, and how these mechanisms affect women’s reproductive health and pregnancy.
Genes encoding proteins involved in bacterial secretion systems play an important role in pathogenicity53. Thus, we examined all genes predicted to encode proteins involved in bacterial secretion in the database. We observed that 81% (71/91) of the predicted genes encoding secreted proteins that were far more transcriptionally abundant in the preterm cohort were from taxa identified as associated with PTB in our 16S rRNA analyses . The detected differences between the PTB and TB samples likely reflect the elevated proportional abundance of these taxa in PTB samples. Further studies are needed to determine whether these genes may contribute to mechanisms by which components of the vaginal microbiome may mediate or cause pathology or PTBs.
Host cytokine expression in PTB
Of the nine cytokine levels examined in the present study (interleukin (IL)-1β, IL-6, IL-8, eotaxin, tumor necrosis factor (TNF)-α, IL-17A, macrophage inflammatory protein (MIP)-1β, interferon-γ-induced protein (IP)-10/chemokine ligand (CXCL)10, RANTES (regulated on activation, normal T cell expressed and secreted)), four (eotaxin, IL-1β, IL-6 and MIP-1β) were greatly increased in PTB relative to TB samples (false discovery rate (FDR)-adjusted P < 0.05 for each), consistent with previous reports of elevated IL-1, IL-6, MIP-1, IP10/CXCL10 and other proinflammatory cytokines associated with PTB in blood, amniotic fluid or cervical–vaginal lavage samples54. For further examination of the role of cytokines in the progression of pregnancy to PTB, we performed an integrative sparse canonical correlation analysis (sCCA) to assess the association of specific bacterial taxa with the abundance levels of nine key cytokines. For each participant, the sample corresponding to the earliest gestational age per trimester was characterized. In women who delivered at term (Fig.3a), we observed a strong negative correlation between L. crispatus and several taxa associated with dysbiosis and PTB (for example, G. vaginalis, Prevotella cluster 2, S. amnii and, to a lesser extent, TM7-H1), as well as with the analyzed cytokines. The analyzed cytokines, which are largely proinflammatory, were loosely correlated both with each other and with taxa associated with dysbiosis and PTB. Notably, IP-10/CXCL10, which functions to induce chemotaxis of immune cells and promotes apoptosis, cell growth and angiostasis, and is generally considered to be proinflammatory55, was positively correlated with L. iners. This association was previously reported in the reproductive tracts of non-pregnant women from Kenya56. In contrast, in women who went on to experience PTB (Fig.3b), the proinflammatory cytokines and dysbiotic taxa (for example, A. vaginae, G. vaginalis and Megasphaera type 1) formed a tighter cluster, indicating a stronger positive correlation, but IP-10/CXCL10 did not correlate with L. iners. Furthermore, BVAB1 was negatively correlated with IP-10/CXCL10 in these samples.
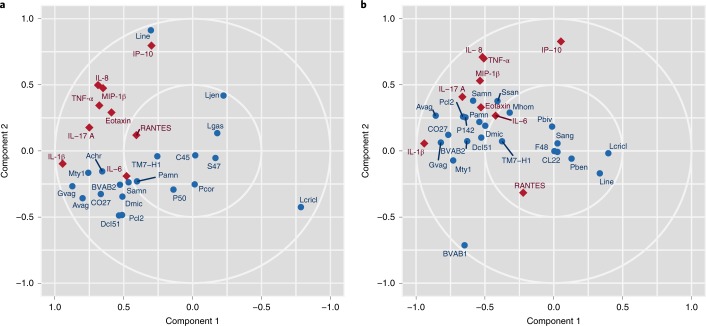
Fig.3 Sparse canonical correlation analysis.
a,b, Cytokine abundance in vaginal samples from women who experienced TB (n = 90) (a) or PTB (n = 41) (b) were subjected to an integrative sCCA using log-transformed cytokine levels and log-transformed taxonomic profiling data (see Methods). Blue circles represent bacterial taxa and red diamonds represent cytokines. Note that the component 1 axis for the TB sCCA (left) has been reversed for effective visual comparison with PTB sCCA. Note that, in sCCA analysis, factors (cytokines or microbial taxa) that are clustered tightly are highly correlated, and factors that are distant from each other are inversely correlated.
Cross-study comparisons of the vaginal microbiome and preterm birth
Several recent studies25,27,28,30 generally reported limited correlation between the composition of the vaginal microbiome and PTB in cohorts of African descent. We compared the distributions of distinct candidate taxa for PTB risk across four studies25,27,30 of the vaginal microbiome in cohorts of pregnant women with predominantly African ancestry, including the MOMS-PI PTB study, with a harmonized reanalysis of the raw 16S rRNA sequencing reads. There were non-trivial differences in every technical aspect of study design including sample collection, DNA extraction, PCR primers and conditions, sequencing platform, data quality and deposit which precluded an integrated analysis of these datasets. Moreover, each of these studies varied markedly in cohort demographics, inclusion and exclusion criteria, and even the definition of PTB. PTB is understood to be a syndrome with many underlying causes9. Ascending infection of microbes from the vagina likely plays a causative role in some subtypes of PTB, but likely does not play a contributing role in all PTB. Thus, there were considerable differences in the PTB case cohorts, as originally published in terms of the distribution of gestational age at delivery, the percentage of women who had a non-medically indicated spontaneous PTB (37.5–100%), preterm premature rupture of the membranes (12.5–57.8%), treatment with progesterone (0–100%) or a history of PTB (29.1–100%). Our attempts to aggregate data from different studies highlight the opportunities for harmonization to obtain comparable data across studies.
The present study confirms an association between Sneathia sanguinegens and PTB, which was reported as significant (P < 0.05) before adjustment for multiple testing by Romero et al.30. Although there were only five spontaneous PTB cases reanalyzed in the Stout et al. cohort27, and only ten in the reanalyzed Callahan et al.25 cohort, we observed high concordance in the directionality of differences in abundance levels of preterm and term groups between these cohorts and the present study.We were also able to confirm that BVAB1, Megasphaera phylotype 1 and Sneathia species were elevated in a preterm cohort, which Nelson et al.26 previously reported as related to an increased risk for PTBs among women reporting a prior preterm delivery. The Nelson et al.26 study used quantitative PCR rather than 16S rRNA profiling, and the study was thus not included in the harmonized reanalysis. We also confirmed that L. iners and G. vaginalis, which were identified as vaginal microbial signatures associated with PTB in low-risk cohorts25, did not generalize to cohorts of African ancestry.
Discussion
From a subset of 597 of the 1,572 pregnancies longitudinally sampled for the MOMS-PI study, we have generated omics data from more than 12,000 samples in one of the largest and most comprehensive multi-omic studies published to date. In addition, our analyses of longitudinal omics data from vaginal samples from 45 women who delivered preterm and 90 controls showed a signature of PTB in a cohort of women of predominantly African ancestry, including several taxa that have previously been implicated in adverse outcomes of pregnancy, including premature delivery22,26,57–59, in addition to taxa that have not been previously linked to adverse pregnancy outcomes. Women of African ancestry have a greatly increased risk of PTB compared with women of European ancestry60. Previous studies16,21 have shown that carriage of L. crispatus, which is negatively associated with PTB, is more prevalent in women of European ancestry, and BVAB1, which is positively associated with PTBs, is more common in women of African ancestry. Thus, our findings are consistent with a proposed framework in which there is a spectrum of vaginal microbiome states linked to risk for PTB, and that these states vary across populations.
Our longitudinal modeling showed taxa associated with PTB tended to decrease in abundance in the vaginal environment throughout pregnancy, particularly in women of African ancestry. This finding is consistent with previous observations that pregnancy is associated with reduced carriage of bacterial vaginosis-associated organisms21,27,57. Considering that an adverse pregnancy outcome may be caused by ascension of pathogenic microbes, this trend suggests that the microbiome composition early in pregnancy may be most useful in the prediction of adverse outcomes. In a complementary analysis of an ethnically diverse cohort of women who delivered at term or early term in the MOMS-PI TB study, we show differences in the longitudinal dynamics of the microbiome in women of African ancestry compared with women of European ancestry42. In the present study, we developed a proof-of-concept model that suggests the presence of BVAB1, Prevotella cluster 2, S. amnii and TM7-H1 early in pregnancy may be useful for prediction of risk for PTB, particularly in high-risk populations. It is possible that BVAB1, Prevotella cluster 2, S. amnii and TM7-H1, and other taxa may have roles in the causation of PTB. As BVAB1 and TM7-H1 had not been cultivated or genetically characterized, we assembled their genomes from MGS data to search for clues to their pathogenic potential. We previously characterized the genome of S. amnii, identified potential cytotoxin genes and showed that cultured bacteria kill eukaryotic cells in vitro61. Although culture is not available for BVAB1 or TM7-H1, genomic factors identified in their genomes can now be genetically amplified and recombined into heterologous reporter systems and tested for pathogenic activity. Our MTS and MGS analyses supported the microbial signatures identified using 16S rRNA profiles.
Analysis of vaginal cytokine data from the MOMS-PI PTB study is consistent with previous findings showing that bacterial taxa generally associated with dysbiosis are highly correlated with expression of proinflammatory cytokines, which may play a role in the induction of labor. Labor is associated with proinflammatory cytokine expression, and premature labor can be induced by host inflammatory responses. We observed that vaginal IP-10/CXCL10 levels were inversely correlated with BVAB1 in PTB, inversely correlated with L. crispatus in TB and positively correlated with L. iners in TB, suggesting complex host–microbiome interactions in pregnancy.
Our findings contribute to an understanding of how microbial markers for PTB vary across populations. Vaginal microbiome composition as a whole and carriage rates of specific microbial taxa vary dramatically across populations, and thus it is not unexpected that the importance of relevant markers differs accordingly. Further studies are needed to determine whether the signatures of PTB reported in the present study replicate in other cohorts of women of African ancestry, to examine whether the observed differences in vaginal microbiome composition between women of different ancestries has a direct causal link to the ethnic and racial disparities in PTB rates, and to establish whether population-specific microbial markers can be ultimately integrated into a generalizable spectrum of vaginal microbiome states linked to the risk for PTB. Taken together, our data suggest that, coupled with other clinical and possibly genetic factors, microbiome-associated taxonomic, metabolic and immunologic biomarkers may be useful in defining the risk of PTB, and that this risk might be assessed early in pregnancy.
References
1. Blencowe H, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet. 2012;379:2162–2172. doi: 10.1016/S0140-6736(12)60820-4.
2. Liu L, et al. Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet. 2016;388:3027–3035. doi: 10.1016/S0140-6736(16)31593-8.
3. Marret S, et al. Neonatal and 5-year outcomes after birth at 30–34 weeks of gestation. Obstet. Gynecol. 2007;110:72–80. doi: 10.1097/01.AOG.0000267498.95402.bd.
4. Wolke D, Eryigit-Madzwamuse S, Gutbrod T. Very preterm/very low birthweight infants’ attachment: infant and maternal characteristics. Arch. Dis. Child. Fetal Neonat. Ed. 2014;99:F70–F75. doi: 10.1136/archdischild-2013-303788.
5. Behrman, R. E. & Butler, A. S. (eds), for Institute of Medicine Committee on Understanding Premature Birth and Assuring Healthy Outcomes. Societal Costs of Preterm Birth (National Academies Press, 2007).
6. Strauss JF, et al. Spontaneous preterm birth: advances toward the discovery of genetic predisposition. Am. J. Obstet. Gynecol. 2018;218:294–314.e2. doi: 10.1016/j.ajog.2017.12.009.
7. Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4.
8. Donders GG, et al. Predictive value for preterm birth of abnormal vaginal flora, bacterial vaginosis and aerobic vaginitis during the first trimester of pregnancy. Br. J. Obstet. Gynaecol. 2009;116:1315–1324. doi: 10.1111/j.1471-0528.2009.02237.x.
9. Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345:760–765. doi: 10.1126/science.1251816.
10. Lamont RF. Infection in the prediction and antibiotics in the prevention of spontaneous preterm labour and preterm birth. Br. J. Obstet. Gynaecol. 2003;110(Suppl 20):71–75. doi: 10.1046/j.1471-0528.2003.00034.x.
11. Lockwood CJ. Predicting premature delivery—no easy task. N. Engl. J. Med. 2002;346:282–284. doi: 10.1056/NEJM200201243460412.
12. Fredricks DN, Fiedler TL, Thomas KK, Oakley BB, Marrazzo JM. Targeted PCR for detection of vaginal bacteria associated with bacterial vaginosis. J. Clin. Microbiol. 2007;45:3270–3276. doi: 10.1128/JCM.01272-07.
13. Sobel JD. Bacterial vaginosis. Annu. Rev. Med. 2000;51:349–356. doi: 10.1146/annurev.med.51.1.349.
14. Bradshaw CS, Sobel JD. Current treatment of bacterial vaginosis: limitations and need for innovation. J. Infect. Dis. 2016;214:S14–S20. doi: 10.1093/infdis/jiw159.
15. Chavoustie SE, et al. Experts explore the state of bacterial vaginosis and the unmet needs facing women and providers. Int. J. Gynaecol. Obstet. 2017;137:107–109. doi: 10.1002/ijgo.12114.
16. Ravel J, et al. Vaginal microbiome of reproductive-age women. Proc. Natl Acad. Sci. USA. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107.
17. Ma B, Forney LJ, Ravel J. Vaginal microbiome: rethinking health and disease. Annu. Rev. Microbiol. 2012;66:371–389. doi: 10.1146/annurev-micro-092611-150157.
18. MacIntyre DA, et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 2015;5:8988. doi: 10.1038/srep08988.
19. Martin DH, Marrazzo JM. The vaginal microbiome: current understanding and future directions. J. Infect. Dis. 2016;214(Suppl 1):S36–S41. doi: 10.1093/infdis/jiw184.
20. Zhou X, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007;1:121–133. doi: 10.1038/ismej.2007.12.
21. Fettweis JM, et al. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology. 2014;160:2272–2282. doi: 10.1099/mic.0.081034-0.
22. Brown RG, et al. Vaginal dysbiosis increases risk of preterm fetal membrane rupture, neonatal sepsis and is exacerbated by erythromycin. BMC Med. 2018;16:9. doi: 10.1186/s12916-017-0999-x.
23. Kindinger LM, et al. The interaction between vaginal microbiota, cervical length, and vaginal progesterone treatment for preterm birth risk. Microbiome. 2017;5:6. doi: 10.1186/s40168-016-0223-9.
24. DiGiulio DB, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl Acad. Sci. USA. 2015;112:11060–11065. doi: 10.1073/pnas.1502875112.
25. Callahan BJ, et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl Acad. Sci. USA. 2017;114:9966–9971. doi: 10.1073/pnas.1705899114.
26. Nelson DB, et al. Early pregnancy changes in bacterial vaginosis-associated bacteria and preterm delivery. Paediatr. Perinat. Epidemiol. 2014;28:88–96. doi: 10.1111/ppe.12106.
27. Stout MJ, et al. Early pregnancy vaginal microbiome trends and preterm birth. Am. J. Obstet. Gynecol. 2017;217:356.e1–356.e18. doi: 10.1016/j.ajog.2017.05.030.
28. Nelson DB, Shin H, Wu J, Dominguez-Bello MG. The gestational vaginal microbiome and spontaneous preterm birth among nulliparous African American women. Am. J. Perinatol. 2016;33:887–893. doi: 10.1055/s-0036-1584581.
29. Han YW, Shen T, Chung P, Buhimschi IA, Buhimschi CS. Uncultivated bacteria as etiologic agents of intra-amniotic inflammation leading to preterm birth. J. Clin. Microbiol. 2009;47:38–47. doi: 10.1128/JCM.01206-08.
30. Romero R, et al. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome. 2014;2:18. doi: 10.1186/2049-2618-2-18.
31. Tabatabaei N, et al. Vaginal microbiome in early pregnancy and subsequent risk of spontaneous preterm birth: a case-control study. Br. J. Obstet. Gynaecol. 2018;126:349–358. doi: 10.1111/1471-0528.15299.
32. Son K-A, et al. Prevalence of vaginal microorganisms among pregnant women according to trimester and association with preterm birth. Obstet. Gynecol. Sci. 2018;61:38–47. doi: 10.5468/ogs.2018.61.1.38.
33. Haque MM, Merchant M, Kumar PN, Dutta A, Mande SS. First-trimester vaginal microbiome diversity: a potential indicator of preterm delivery risk. Sci. Rep. 2017;7:16145. doi: 10.1038/s41598-017-16352-y.
34. Freitas AC, et al. The vaginal microbiome of pregnant women is less rich and diverse, with lower prevalence of Mollicutes, compared to non-pregnant women. Sci. Rep. 2017;7:9212. doi: 10.1038/s41598-017-07790-9.
35. Subramaniam A, et al. Vaginal microbiota in pregnancy: evaluation based on vaginal flora, birth outcome, and race. Am. J. Perinatol. 2016;33:401–408.
36. Hyman RW, et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 2014;21:32–40. doi: 10.1177/1933719113488838.
37. Stafford GP, et al. Spontaneous preterm birth is associated with differential expression of vaginal metabolites by lactobacilli-dominated microflora. Front. Physiol. 2017;8:615. doi: 10.3389/fphys.2017.00615.
38. Jefferson KK, et al. Relationship between vitamin D status and the vaginal microbiome during pregnancy. J. Perinatol. 2019;39:824–836. doi: 10.1038/s41372-019-0343-8.
39. Brown RG, et al. Prospective observational study of vaginal microbiota pre- and post-rescue cervical cerclage. Br. J. Obstet. Gynaecol. 2019;126:916–925. doi: 10.1111/1471-0528.15600.
40. Borgdorff H, et al. The association between ethnicity and vaginal microbiota composition in Amsterdam, the Netherlands. PloS ONE. 2017;12:e0181135. doi: 10.1371/journal.pone.0181135.
41. The Integrative Human Microbiome Project. Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16:276–289. doi: 10.1016/j.chom.2014.08.014.
42. Serrano Myrna G., et al.Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nature Medicine. 2019;25(6):1001–1011. doi: 10.1038/s41591-019-0465-8.
43. Leal M, et al. Burden of early-term birth on adverse infant outcomes: a population-based cohort study in Brazil. BMJ Open. 2017;7:e017789. doi: 10.1136/bmjopen-2017-017789.
44. Murray SR, et al. Long term cognitive outcomes of early term (37–38 weeks) and late preterm (34–36 weeks) births: a systematic review. Wellcome Open Res. 2017;2:101. doi: 10.12688/wellcomeopenres.12783.1.
45. Boyle EM, et al. Effects of gestational age at birth on health outcomes at 3 and 5 years of age: population based cohort study. BMJ. 2012;344:e896. doi: 10.1136/bmj.e896.
46. Brooks J. Paul, Buck Gregory A., Chen Guanhua, Diao Liyang, Edwards David J., Fettweis Jennifer M., Huzurbazar Snehalata, Rakitin Alexander, Satten Glen A., Smirnova Ekaterina, Waks Zeev, Wright Michelle L., Yanover Chen, Zhou Yi-Hui. Changes in vaginal community state types reflect major shifts in the microbiome. Microbial Ecology in Health and Disease. 2017;28(1):1303265. doi: 10.1080/16512235.2017.1303265.
47. Fettweis JM, et al. Species-level classification of the vaginal microbiome. BMC Genom. 2012;13(Suppl 8):S17.
48. Romero R, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. 2014;2:4. doi: 10.1186/2049-2618-2-4.
49. Walther-António MRS, et al. Pregnancy’s stronghold on the vaginal microbiome. PloS ONE. 2014;9:e98514. doi: 10.1371/journal.pone.0098514.
50. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015.
51. Aldunate M, et al. Antimicrobial and immune modulatory effects of lactic acid and short chain fatty acids produced by vaginal microbiota associated with eubiosis and bacterial vaginosis. Front. Physiol. 2015;6:164. doi: 10.3389/fphys.2015.00164.
52. He X, et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl Acad. Sci. USA. 2015;112:244–249. doi: 10.1073/pnas.1419038112.
53. Green, E. R. & Mecsas, J. In Virulence Mechanisms of Bacterial Pathogens 5th edn (eds Kudva, I. et al.) 215–239 (2016).
54. Wei S-Q, Fraser W, Luo Z-C. Inflammatory cytokines and spontaneous preterm birth in asymptomatic women: a systematic review. Obstet. Gynecol. 2010;116:393–401. doi: 10.1097/AOG.0b013e3181e6dbc0.
55. Liu M, et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011;22:121–130.
56. Jespers V, et al. A longitudinal analysis of the vaginal microbiota and vaginal immune mediators in women from sub-Saharan. Afr. Sci. Rep. 2017;7:11974. doi: 10.1038/s41598-017-12198-6.
57. Freitas AC, Bocking A, Hill JE, Money DM. VOGUE Research Group. Increased richness and diversity of the vaginal microbiota and spontaneous preterm birth. Microbiome. 2018;6:117. doi: 10.1186/s40168-018-0502-8.
58. Baldwin EA, et al. Persistent microbial dysbiosis in preterm premature rupture of membranes from onset until delivery. PeerJ. 2015;3:e1398. doi: 10.7717/peerj.1398.
59. Paramel Jayaprakash T, et al. High diversity and variability in the vaginal microbiome in women following preterm premature rupture of membranes (PPROM): a prospective cohort study. PloS ONE. 2016;11:e0166794. doi: 10.1371/journal.pone.0166794.
60. Blencowe H, et al. Born too soon: the global epidemiology of 15 million preterm births. Reprod. Health. 2013;10:S2. doi: 10.1186/1742-4755-10-S1-S2.
61. Harwich MD, Jr, et al. Genomic sequence analysis and characterization of Sneathia amnii sp. nov. BMC Genom. 2012;13(Suppl 8):S4. doi: 10.1186/1471-2164-13-S8-S4.


